
Genética de la infertilidad masculina
Nuestro especialista en infertilidad masculina:
Está claro que una proporción significativa de hombres infértiles con azoospermia y oligospermia severa tienen una etiología genética para la falla reproductiva. Si bien los recientes avances en las tecnologías de reproducción asistida han sido posibles y prácticos para muchos hombres infértiles con infertilidad masculina severa a los hijos de los padres, también plantearon preocupaciones acerca de transmitir anomalías genéticas a los hijos de estos hombres. La inyección intracitoplasmática de espermatozoides (ICSI) es la técnica más invasiva para la reproducción asistida. ICSI pasa por alto todos los mecanismos fisiológicos relacionados con la fertilización, así como todas las barreras protectoras contra los espermatozoides con defectos genéticos y permitir incluso alterado espermatozoide para fertilizar un ovocito. Dado que los pacientes infértiles con azoospermia no obstructiva son capaces de lograr el embarazo con espermatozoides testiculares recuperados quirúrgicamente, ICSI lleva el riesgo de transmitir tanto enfermedades determinadas genéticamente como la infertilidad genéticamente determinada. Es imprescindible para los clínicos involucrados en el tratamiento de estas parejas para iniciar la evaluación genética y asesoramiento antes de cualquier procedimiento terapéutico.
Todas las células diploides nucleadas humanas contienen 22 pares de autosomas y 1 par de cromosomas sexuales, para un total de 23 pares, que proporciona el normal 46, XX (mujer) y 46, XY (masculino) la configuración cromosómica. Es importante reconocer que la única fuente del cromosoma Y es paternal.
Se estima que el genoma humano contiene genes 50,000-100,000 que codifican proteínas.
Fibrosis quística
La fibrosis quística (FQ) Se ha detectado una mutación genética en hombres con ausencia congénita del conducto deferente, pero sin otra manifestación de CF. La FQ es el trastorno autosómico recesivo fatal más frecuente en la población caucásica con la incidencia de aproximadamente 1 en nacidos vivos 2400. La frecuencia del portador es más alta en 1 en personas 25 en población de ascendencia del norte de Europa. El gen de la FC fue identificado y clonado en 1989 y ha sido denominado el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR). Los estudios genéticos revelaron que 50-83% de los pacientes Congenital BIlateral ABsencia del Vas DEferensCBAVD) tienen al menos una mutación del gen CFTR conocida y que aproximadamente el 10% tiene dos mutaciones CFTR conocidas. La mayoría de los hombres con CBAVD tienen solo una mutación o variante de un gen detectable. Las mutaciones más comunes son D 508 (70%), 5T, R117H, R75Q, G542X, N1303, W1282X, G551A y R347H. En aquellos hombres con mutaciones identificadas en ambos alelos de CFTR, al menos una mutación es leve. Más del 95% de hombres con FQ tienen anomalías en las estructuras derivadas del conducto de Wolff. La ausencia del conducto deferente puede ser el único signo clínico de presentación en estos pacientes. El epidídimo del cuerpo y la cauda puede estar presente, ausente o atróficamente, pero el epidídimo caput está presente invariablemente. Se ha especulado que cuanto más largo sea el remanente de epidídimo, mayor será la tasa de éxito de la aspiración de esperma. CBAVD puede representar leve de la FQ. Hasta el 82% de pacientes con CBAVD tienen al menos una mutación de FQ detectable. La ausencia bilateral congénita de los conductos deferentes se encuentra en 2% de hombres que presentan infertilidad. Es una causa común de azoospermia asociada con un bajo volumen de semen y un pH ácido. La espermatogénesis suele estar intacta, por lo que actualmente no se recomienda la biopsia testicular en estos pacientes. La cantidad suficiente de espermatozoides por lo general se puede recuperar del epidídimo obstruido o de su remanente.
Si un hombre con CBAVD está contemplando la aspiración microquirúrgica del esperma epididimal con ICSI subsecuente, el análisis completo de la mutación del esposo CF es obligatorio para definir el riesgo de pasar a lo largo de CF o CBAVD. Si el cónyuge es negativo para la mutación común, su riesgo de portador de FQ es de 0.4%. Un riesgo para esta pareja de tener un niño con genotipo CF no es más que 0.2%. Si la esposa es portadora, entonces las posibilidades de que el niño tenga genotipo CF es 50%.
Congenital UNilateral ABsencia del Vas DEferensCUAVD) Afecta a 0.5-1% de la población general y rara vez se presenta con infertilidad. Se han reportado mutaciones en el gen de CFTR en 43% de hombres con ausencia unilateral de los conductos deferentes y 47% de hombres sanos con obstrucción idiopática del epidídimo.
Anomalías urogenitales en pacientes con ausencia de vasos deferentes..
La agenesia renal se encontró en 11% de pacientes con CBAVD y 26% de pacientes con CUAVD
Vesículas seminales se encontraron en 55% de los pacientes con CBAVD. Vesícula seminal Ipsilateral estuvo presente sólo en 14% de pacientes con CUAVD.
Evaluación del cariotipo
La anomalía más frecuente del cariotipo en hombres con infertilidad masculina severa es el síndrome de Klinefelter, que afecta a 7-13% de hombres azoospérmicos. La prevalencia del síndrome de Klinefelter se estima en aproximadamente 1 en varones vivos 600. La tríada clásica incluye pequeños testículos firmes, ginecomastia y azoospermia. 90% de todos los pacientes tienen 46, XXY cariotipo y 10% son mosaicos con cariotipo 46XY / 47, XXY. La recuperación de esperma es posible en estos pacientes a pesar de los cambios histológicos severos (hialinización de los túbulos seminíferos e hiperplasia de las células de Leydig) con posterior fertilización y embarazo.
También se sugiere que casi uno de cada veinte hombres oligospermic tenía un cariotipo anormal (principalmente Robertsonian y translocations reciprocal). Por lo tanto, el cariotipo debe considerarse como una parte obligatoria del proceso de cribado previo al tratamiento para todos los hombres remitidos para ICSI.
Microdeleciones del cromosoma Y
Tiepolo y Zufardi en 1976 encontraron la deleción del extremo distal del brazo largo del cromosoma Y. Esta región incluye el locus del factor azoospermia (AZF), que contiene un gen o genes, necesarios para la espermatogénesis. El locus AZF se ha asignado al intervalo de deleción 5-7, una región en la banda q11.23 del cromosoma Y que contiene 5 millones de pares de bases. Hay tres regiones en el intervalo de deleción 5 y 6 donde la mayoría de las supresiones se producen en el hombre infértil, designado como AZFa, AZFb y AZFc. Cada una de estas regiones puede estar asociada con una histología testicular particular. En un estudio se encontraron microdeleciones en el cromosoma Y en 7% de grupo de hombres infértiles no seleccionados, lo que sugiere que las microdeleciones de cromosomas Y constituyen la segunda causa específica más común de infertilidad masculina.
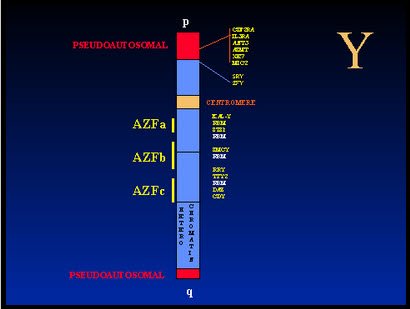
Los genes AZF aún no han sido identificados. Dos familias de genes candidatos se clonaron actualmente a partir del intervalo de deleción 6: RBM (RN / A-BInding MOtif) y DAZ (DMontado en AzOospermia). Los genes DAZ se expresan exclusivamente en la prueba. Las proteínas DAZ parecen estar presentes en las espermátidas tardías y en las colas de los espermatozoides maduros. La ausencia de deleciones detectables en los hombres fértiles normales sugiere una relación de causa a efecto entre las deleciones y la infertilidad. Sin embargo, se demostró que algunos hombres con microdeleciones pueden producir esperma en el eyaculado. Además, los hombres azoospérmicos con deleción del grupo de genes DAZ ocasionalmente son capaces de completar la espermatogénesis. Se especuló que las microdeleciones AZFc son compatibles con la formación de un pequeño número de espermatozoides normales. Cuando se recuperaron espermatozoides testiculares se utilizaron para la reproducción asistida (FIV / ICSI), el desarrollo embrionario normal y el embarazo han sido documentados. El rol funcional de los genes del cromosoma Y en la espermatogénesis quedó por identificar. Se sugiere que las microdeleciones del cromosoma Y ocurren durante la replicación del ADN. Entre los hombres con azoospermia o oligospermia severa, 16% tuvo deleciones. En los hombres con azoospermia sólo la frecuencia de deleciones aumentó a 23%.
Las microdeleciones del cromosoma Y son relativamente frecuentes y su frecuencia aumenta con la gravedad del defecto espermatogénico. Por lo tanto, la prueba de microdeleciones del cromosoma Y está indicada en hombres con azoospermia y oligospermia severa, y en pacientes que consideran la reproducción asistida.
Las microdeleciones del cromosoma Y no pueden predecirse sobre la base de los hallazgos clínicos o de los resultados del análisis del semen. Las pruebas para las microdeleciones del cromosoma Y implican la extracción de ADN de los glóbulos blancos y la amplificación de una región específica del cromosoma Y en la PCR que contiene los pares de cebadores 5-8. Cada par de cebadores amplifica una región específica del cromosoma Y (un sitio marcado con secuencia).
Las anormalidades del cromosoma Y se transmitirán a cualquier niño varón que se produzca después de la reproducción asistida. Dado que los hombres con estos trastornos genéticos suelen ser perfectamente saludables, aparte de la infertilidad, es incierto si alguna otra condición médica estará presente en los descendientes con microdeleciones de cromosomas Y.
El asesoramiento genético es importante antes del tratamiento. La detección de microdeleciones de cromosomas Y puede proporcionar el diagnóstico de infertilidad y permitir que el médico dirija a los pacientes a la reproducción o adopción asistida. Dado que los hijos de hombres con microdeleciones de cromosomas Y concebidos con reproducción asistida serán infértiles, la infertilidad del factor masculino debe ser discutida.
Los datos actuales no sugieren un mayor riesgo de enfermedades genéticamente transmitidas en niños nacidos después de ICSI. Las malformaciones mayores se encontraron en un rango de 2.4%, que es comparable a las cifras conocidas de niños nacidos después de FIV o concepción natural. Sin embargo, los pacientes deben ser aconsejados antes de cualquier tratamiento sobre la base de los datos disponibles sobre el riesgo de defectos cromosómicos transmitidos, aberraciones cromosómicas de novo y problemas de fertilidad a la descendencia.